Ante el exito de los mensajes dedicados al análisis cluster la nueva entrega del manual de R la dedicaremos de nuevo al análisis de agrupamiento. Como es habitual trabajaremos con un ejemplo que podéis desgargaros aquí. Partimos de un archivo de texto delimitado por tabuladores con 46 frutas y la información que disponemos es:
- Nombre
- Intercambio de hidratos de carbono por gramo
- Kilocalorías
- Proteinas
- Grasas
(información obtenida de www.diabetesjuvenil.com)
El primer paso será crear un objeto en R que recoja los datos en el análisis. Para ello vamos a emplear la función read.table que deberá tener los parámetros adecuados al fichero de texto que deseamos leer:
frutas<-read.table("c:\\datos\\alimentos.txt",header=FALSE,sep="\t")
nombres<-c("nombre","inter_hidratos","kcal","proteinas","grasas")
names(frutas)<-nombres
El archivo de texto lo tenemos en una ubicación de nuestra máquina c:\datos, pero lo he subido y también podemos leerlo directamente de la web:
frutas<-read.table(url("https://analisisydecision.es/wp-content/uploads/2009/06/alimentos2.txt"),header=FALSE,sep="\t")
nombres<-c("nombre","inter_hidratos","kcal","proteinas","grasas")
names(frutas)<-nombres
Lo leemos sin cabeceras y como separador indicamos el tabulador con el parámetro de read.table sep=»\t». Ya disponemos del objeto para el análisis. Como vimos en capítulos anteriores el primer paso es crear la matriz de distancias, realizar el cluster con ella y seleccionar el número de grupos. Para crear la matriz de distancias entre observaciones hemos de especificar un método de cálculo, en este punto vamos a aprobechar para comparar 4 métodos de obtención de distancias:
distancias1<-dist(frutas,method="manhattan")
cluster1<-hclust(distancias1)
distancias2<-dist(frutas,method="euclidean")
cluster2<-hclust(distancias2)
distancias3<-dist(frutas,method="maximum")
cluster3<-hclust(distancias3)
distancias4<-dist(frutas,method="canberra")
cluster4<-hclust(distancias4)
#Realizamos la comparativa gráfica
op <- par(mfcol = c(2, 2)) #Nos permite presentar
par(las =1) #el gráfico en 4 partes
plot(cluster1,main="Método Manhatan")
plot(cluster2,main="Distancia euclídea")
plot(cluster3,main="Distancia por máximos")
plot(cluster4,main="Método Camberra")
Disponemos de 4 gráficos en la pantalla que nos permiten comparar los distintos métodos empleados para las distancias. Vemos que tanto la distancia euclídea, como máximos y Manhatan ofrecen resultados parecidos, parece que se forman 4 grupos y las observaciones 41 y 1 son muy distintas al resto. El método Camberra es el que ofrece otros resultados diferentes pero este método es adecuado para datos estandarizados y no es el caso. En nuestro ejemplo vamos a emplear la distancia euclídea por lo que emprearemos el objeto cluster2. Si observamos el dendograma que nos ofrece R de cluster2 parece que podemos formar 4 grupos y hay observaciones muy distintas del resto. Para determinar mejor el número de clusters a seleccionar vamos a emplear el algoritmo PAM (Partitioning Around Medoids):
library(cluster)
paso1<-pam(distancias2,2)
paso2<-pam(distancias2,3)
paso3<-pam(distancias2,4)
paso4<-pam(distancias2,5)
par(mfrow=c(2,2))
plot(paso1)
plot(paso2)
plot(paso3)
plot(paso4)
El resultado de la ejecución de este código lo tenemos en la siguiente figura:
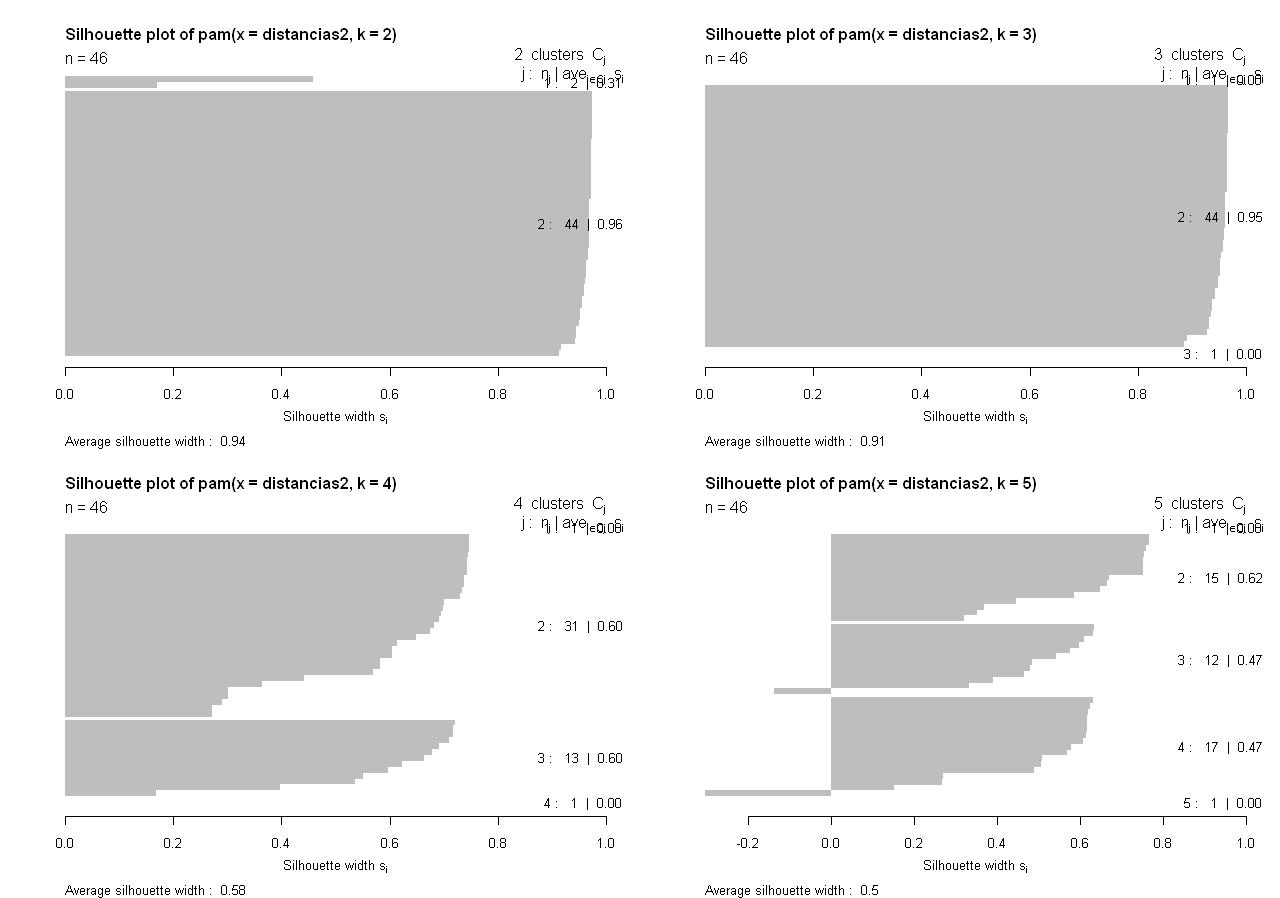
Viendo las 4 siluetas parece más adecuado elegir los k=4 grupos porque son más homogéneos. De todos modos procede un análisis del tamaño de los grupos porque a la vista de las siluetas y los dendogramas anteriores parece que algunas observaciones distorsionan el agrupamiento:
cluster.final<- kmeans(distancias2,3)
cluster.finalsize #Obtenemos el tamaño de los cluster
[1] 13 2 31
cluster.final<- kmeans(distancias2,4)
cluster.finalsize #Obtenemos el tamaño de los cluster
[1] 16 17 2 11
cluster.final<- kmeans(distancias2,5)
cluster.final$size #Obtenemos el tamaño de los cluster
[1] 13 11 17 2 3
Nos quedamos con 4 grupos aunque vemos que uno de ellos tiene sólo dos frutas. Estudiemos como se han agrupado observando las 46 frutas en estudio directamente sobre los datos, para ello creamos un data.frame que sea la unión del objeto frutas y los cluster el análisis final:
cluster.final<- kmeans(distancias2,4)
grupos<-data.frame(frutas)
clus<-as.factor(cluster.final$cluster)
grupos<-cbind(data.frame(frutas),clus)
Para estudiar como se forman los cluster directamente sobre los datos es necesaria una ordenación de los datos. Para ordenar data.frame vamos a emplear el módulo reshape que incluye la función sort_df:
install.packages("reshape")
Vemos que también se ha instalado el paquete plyr porque es necesario para el funcionamiento de reshape. Ahora estamos en disposición de emplear la función sort_df para la ordenación de data.frame:
library(reshape)
grupos<-sort_df(grupos,vars='clus')
grupos
V1 V2 V3 V4 V5 clus
2 ALBARICOQUE 105 41.8 0.84 0.1 1
8 CIRUELAS 91 40.9 1.00 0.1 1
10 FRAMBUESA 125 48.8 1.00 0.8 1
12 GRANADA 133 42.4 1.00 0.1 1
***
nombres<-c("nombre","inter_hidratos","kcal","proteinas","grasas","clus")
names(grupos)<-nombres
Observemos que el data.frame no nos ha respetado los nombres por ese motivo se emplea de nuevo names.
Es recomendable para conocer el funcionamiento del agrupamiento realizar un análisis de las variables dentro de cada cluster. Para ello vamos a emplear la función aggregate que nos realiza sumarizaciones por factores de un data.frame:
aggregate(gruposinter_hidratos,list(gruposclus),mean)
Group.1 x
1 1 102.5294
2 2 509.5000
3 3 168.8182
4 4 54.6250
aggregate(gruposkcal,list(gruposclus),mean)
Group.1 x
1 1 41.97059
2 2 1453.95000
3 3 46.44545
4 4 40.40625
aggregate(gruposproteinas,list(gruposclus),mean)
Group.1 x
1 1 0.637647
2 2 6.000000
3 3 1.263636
4 4 0.362500
aggregate(gruposgrasas,list(gruposclus),mean)
Group.1 x
1 1 0.6588235
2 2 53.2500000
3 3 0.4727273
4 4 0.1437500
Vemos que las frutas del cluster 2 (aguacate y ruibardo) tienen un aporte calórico, protéico y de grasas muy alto; equivalen a comer carne. El grupo 3 destaca por hidratos y proteinas, todo lo contrario que el grupo 4. El grupo 1 se aleja de los grupos 3 y 4 debido a su aporte en hidratos y grasas.
Con este nuevo ejemplo damos por finalizados los capítulos dedicados al análisis cluster. Estos casos están teniendo un gran éxito en la web, además, por el tiempo de permanencia en ellos deben de estar siendo muy útiles. Por supuesto para cualquier duda o sugerencia estamos a vuestra disposición.
Oye, ¿por qué sólo usas PAM para calcular el número «óptimo» de grupos y, luego, para obtenerlos, te decantas por kmeans?
Aunque kmeans se utilice mucho y sea el algoritmo por defecto, a sus casi sesenta años de edad está mandado a recoger. ¿Por qué no usar PAM directamente?
Yo creo que estos artículos se centran más en una visión del proceso general y como se trabaja en R que en un algoritmo concreto. Quizá cabría un artículo por separado y más específico discutiendo distintas alternativas robustas al algoritmo de k-medias, que hay unas cuantas. Entiendo que la pesona que conozca el PAM probablemente no necesite este tipo de artículos para familiarizarse con el análisis cluster en R.
Al final lo que pretendo con estos mensajes es dar a conocer la herramienta. Es una labor más divulgativa, más de propagar el uso de R que científica, para eso os tengo a vosotros los verdaderamente expertos.
Una pregunta, una vez realizas todo el proceso y ya divides a los individuos en grupos cómo puedes representarlos en forma de árbol (los n grupos) o con otro gráfico para poder visualizarlo mejor???
Un saludo y muchas gracias por este curso introductorio de R
Un cordial saludo.
Quería hacer una consulta. Una vez que hemos realizado todo el proceso y tenemos el grupo al que pertenece cada individuo de nuestro fichero, cómo podemos hacer para representarlo en forma de árbol para poder visualizar los grupos en un solo gráfico???
Gracias
Como se hace uuna curva ROC en R?
muchisimas gracias!! trabajo con R y me ha servido de mucho tu ayuda.
Un saludo
Buenos dias queria saber en R-commander NO desde la CONSOLA de R sino desde R-comammander en que menu se mete uno para hacer una curva ROC?
Existe algun programa para la Estimacion de la funcion de supervivencia con el metodo del vecino mas cercano y el kernell de la funcion bivariada?
Hola, imagino que deseas emplear el método de Kaplan-Meier basado en vecinos. Buscando he encontrado el paquete survivalROC:
http://cran.r-project.org/web/packages/survivalROC/survivalROC.pdf
De todos modos ponte en contacto con la lista de R en España y habrá gente que trabaje más habitualmente con esos métodos.
Muchísimas gracias por esta aportación. Muy bien explicado. Me ha sido de gran utilidad.
Dendrograma viene del griego dendros = árbol, y grama = representación gráfica; dendrograma = gráfico de árbol. ¿De dónde viene “dendograma”?, por favor.
Hola ..no he logrado realizar cluster con distancia jaccard–me sale error
garcias
Hola, ¿puede ser que no estés pasando un objeto distancia?
#install.packages(«philentropy»)
library(philentropy)
frutas2<- frutas[,-1] distancias<-as.dist(distance(frutas2,method="jaccard")) cluster1<-hclust(distancias) con as.dist ya creas el objeto que necesita la función hclust. Mira a ver si van por ahí los tiros. Saludos.
hola, tengo un problema no he podido hacer que en el gráfico salga el nombre en lugar del numero
Hola!! ¿Es posible realizar un cluster creando tu la distancia, en vez de utilizar la distancia euclídea o manhattan… etc.?
En caso afirmativo, cómo sería el codigo de R?
Hola
Es posible que este comando «MorphDistMatrix» haya dejado de funcionar?
Solía estar en el packete Claddis
Conocen alguna comando parecido para calcular la matriz de distancia usando una matriz de datos filogenéticos?
Muchas Gracias
Hola, es una pregunta muy específica. ¿Conoces la lista de usuarios de R en español http://r-es.org/2016/03/07/la-lista-de-correo-r-help-es/ ? Creo que hay biólogos que pueden ayudarte. Saludos.
Ok, muchas gracias por responder